Session 1¶
Implementing a 2D simulation of Active Brownian Particles (ABP) in Python.¶
Overview of the problem¶
Description of the model¶
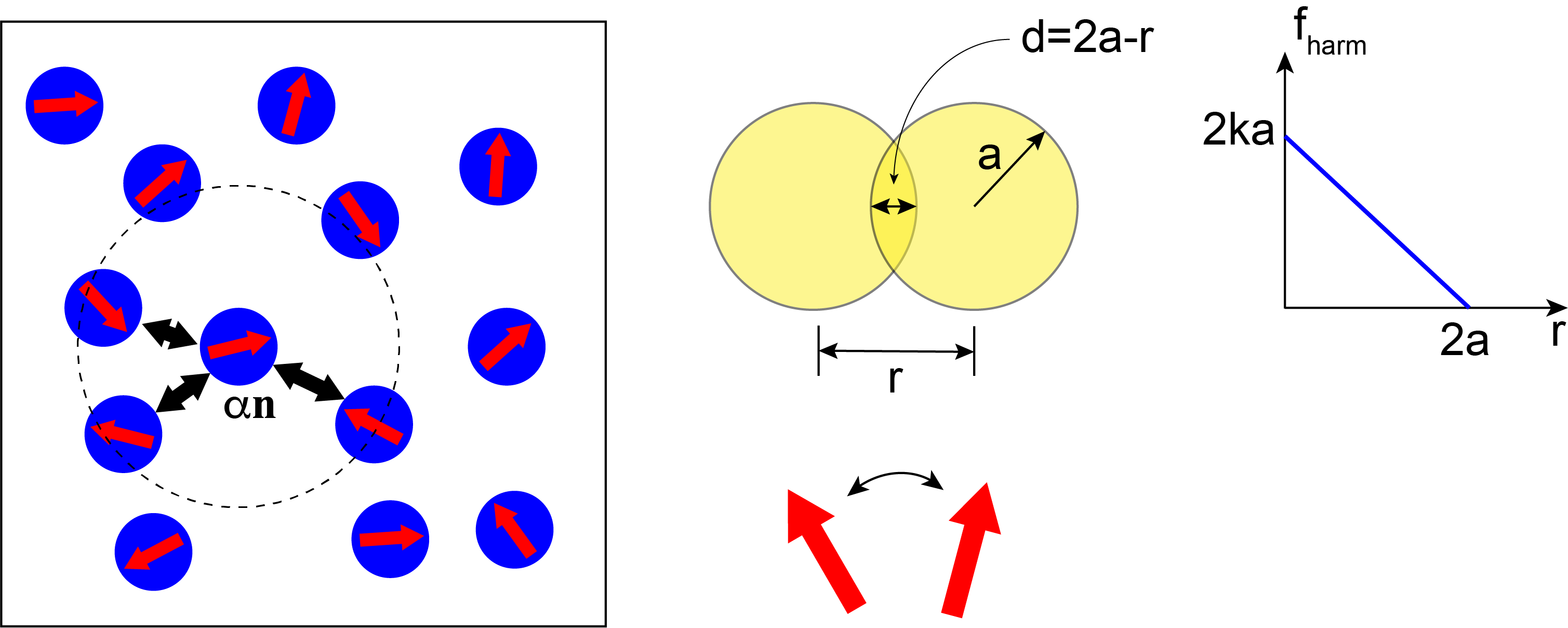
Let us consider a two-dimensional system consisting of $N$ identical disks of radius $a$. The instantaneous position of disk $i$ is given by the radius vector $\mathbf{r}_i = x_i\mathbf{e}_x+y_i\mathbf{e}_y$. In addition, each disk is polar and its polarity is described by a vector $\mathbf{n}_i = \cos(\vartheta_i)\mathbf{e}_x + \sin(\vartheta_i)\mathbf{e}_y$, where $\vartheta_i$ is angle between $\mathbf{n}_i$ and the $x-$axis of the laboratory reference frame. It is immediately clear that $\left|\mathbf{n}_i\right| = 1$.
Disks are assumed to be soft, i.e., they repel each other if they overlap. The interaction force is therefore short-range and, for simplicity, we assume it to be harmonic, i.e., the force between disks $i$ and $j$ is \begin{equation} \mathbf{F}_{ij} = \begin{cases} k\left(2a - r_{ij}\right)\hat{\mathbf{r}}_{ij} & \text{if } r_{ij} \le 2a \\ 0 & \text{otherwise} \end{cases},\label{eq:force} \end{equation} where $k$ is the spring stiffness constant, $\mathbf{r}_{ij} = \mathbf{r}_i - \mathbf{r}_j$, $r_{ij} = \left|\mathbf{r}_{ij}\right|$ is the distance between particles $i$ and $j$ and $\hat{\mathbf{r}}_{ij} = \frac{\mathbf{r}_{ij}}{r_{ij}}$.
Furthermore, overlapping disk are assumed to experience torque that acts to align their polarities. Torque on disk $i$ due to disk $j$ is \begin{equation} \boldsymbol{\tau}_{ij} = \begin{cases} J\mathbf{n}_i\times\mathbf{n}_j & \text{if } r_{ij} \le 2a \\ 0 & \text{otherwise} \end{cases}\label{eq:torque}, \end{equation} where $J$ is the alignment strength. Note that since we are working in two dimensions, $\boldsymbol{\tau}_{ij} = \tau_{ij}\mathbf{e}_z$. It is easy to show that $\tau_{ij} = -J\sin\left(\vartheta_i-\vartheta_j\right)$.
Finally, each disk is self-propelled along the direction of its vector with a force \begin{equation} \mathbf{F}_i = \alpha \mathbf{n}_i,\label{eq:spforce} \end{equation} where $\alpha$ is the magnitude of the self-propulsion force.
Key ingredients of the model are show in the figure below:
Equations of motion¶
In the overdamped limit, all inertial effects are neglected and the equations of motion become simply force (and torque) balance equations. For the model defined above we have, \begin{eqnarray} \dot{\mathbf{r}}_i & = & v_0 \mathbf{n}_i + \frac{1}{\gamma_t}\sum_j \mathbf{F}_{ij} + \boldsymbol{\xi}^{t}_{i}\\ \dot{\vartheta}_i & = & -\frac{1}{\tau_r}\sum_j\sin\left(\vartheta_i-\vartheta_j\right) + \xi_i^r.\label{eq:motion_theta} \end{eqnarray} In previous equations we introduced the translational and rotational friction coefficients, $\gamma_t$ and $\gamma_r$, respectively and defined $v_0 = \frac{\alpha}{\gamma_t}$ and $\tau_r = \frac{\gamma_r}{J}$. Note that $v_0$ has units of velocity (hence we interpret it as the self-propulsion speed) while $\tau_r$ has units of time (hence we interpret it as the polarity alignment time scale). $\boldsymbol{\xi}_i^t$ is the white noise, which we here assume to be thermal noise (in general, this assumption is not required for a system out of equilibrium), i.e., $\langle\xi_{i,\alpha}\rangle=0$ and $\langle \xi_{i,\alpha}^t(t)\xi_{j,\beta}^t(t^\prime)\rangle = 2\frac{k_BT}{\gamma_t}\delta_{ij}\delta_{\alpha\beta}\delta(t-t^\prime)$, with $\alpha, \beta \in \{x, y\}$ and $T$ being temperature. Similarly, $\xi_i^r$ is the rotational noise, with $\langle\xi_i^r\rangle = 0$ and $\langle \xi_i^r(t)\xi_j^r(t^\prime)\rangle = 2D_r\delta_{ij}\delta(t-t^\prime)$, where we have introduced the rotational diffusion coefficient $D_r$.
Overview of a particle-based simulation¶
A typical particle-based simulation workflow consists of three steps:
- Creating the initial configuration;
- Executing the simulation;
- Analyzing the results.
The standard workflow is: step 1 feeds into step 2, which, in turn feeds into step 3.
Creating the initial configuration¶
In the first step, we need to generate the system we would like to study, i.e. we need to create the initial configuration for our simulation. Sometimes, as it is the case here, this is a fairly simple task. However, creating a proper initial configuration can also be a challenging task in its own right that requires a set of sophisticated tools to do. A good example would be setting up a simulation of several complex biomolecules.
Regardless of the complexity, typically the result of this step is one (or several) text (sometimes binary) files that contain information such as the size of the simulation box, initial positions and velocities of all particles, particle connectivities, etc.
In this tutorial, we will use a set of simple Python scripts (located in Python/pymd/builder directory) to create the initial configuration that will be saved as a single JSON file.
For example, let us build the initial configuration with a square simulation box of size $L=50$ with the particle number density $\phi=\frac{N}{L^2}=0.4$. We also make sure that centers of no two particles less than $a=1$ apart. Note that below we will assume that particles have unit radius. This means that our initial configuration builder allowed for significant overlaps between particles. In practice this is not a problem since we will be using soft-core potential and even large overlaps do not lead to excessive force. If one is to consider a system where particles interact via a potential with a strong repulsive component (e.g., the Lennard-Jones potential) the initial configuration would have to be constructed more carefully.
from pymd.builder import *
phi = 0.4
L = 50
a = 1.0
random_init(phi, L, rcut=a, outfile='init.json')
A simple inspection of the file init.json confirms that $N=1000$ particles have been created in the simulation box of size $50\times50$.
Executing the simulation¶
This step (i.e., step 2) is the focus of this tutorial. Conceptually, this step is straightforward - we need to write a program that solves the set equations for motion for a collection of $N$ particles placed in the simulation box. In practice, this is technically the most challenging step and some of the most advanced particle based codes can contain hundreds of thousands of lines of code.
The problem is that equations of motions are coupled, i.e., we are tasked with solving the N-body problem. Therefore, any naive implementation would be so slow that it would not be possible to simulate even the simplest systems within any reasonable time. For this reason, even the most basic particle based codes have to implement several algorithms that make the problem tractable. Furthermore, any slightly more complex problem would require parallelization, which is the topic of the last session in this tutorial.
Analyzing the results¶
Once the simulation in the step 2 has been completed, we need to perform a series of measurements to extract as much physical information about our system as possible. Although sometimes some basic analysis can be done by the main simulation code (i.e., in step 2), this is typically done a posteriori with a separate set of tools. Technically speaking, the main simulation code produces a file (or a set of files), often referred to as trajectories, that are loaded into the analysis code for post-processing.
One of the key steps in the post-processing stage is visualization, e.g. making a movie of the time evolution of the system. Therefore, most simulation codes are able to output the data in multiple file formats for various types of post-processing analysis.
In this tutorial we will output the data in JSON and VTP formats. VTP files can be directly visualized with the powerful Paraview software package.
Writing a particle based simulation in Python¶
Here we outline the key parts of a modern implementation of a particle-based simulation. We use Python for its simple syntax and powerful data structures.
Periodic boundary conditions¶
Before we dive deep into the inner workings of a particle-based code, let us quickly discuss the use of the periodic boundary conditions (PBC).
Even the most advanced particle-based simulations can simulate no more than several million particles. Most simulations are far more modest in size. A typical experimental system usually contains far more particles than it would be possible to simulate. Therefore, most simulations study a small section of an actual system and extrapolate the results onto the experimentally relevant scales. It is, therefore, important to minimize the finite size effects. One such approach is to use the periodic boundary conditions.
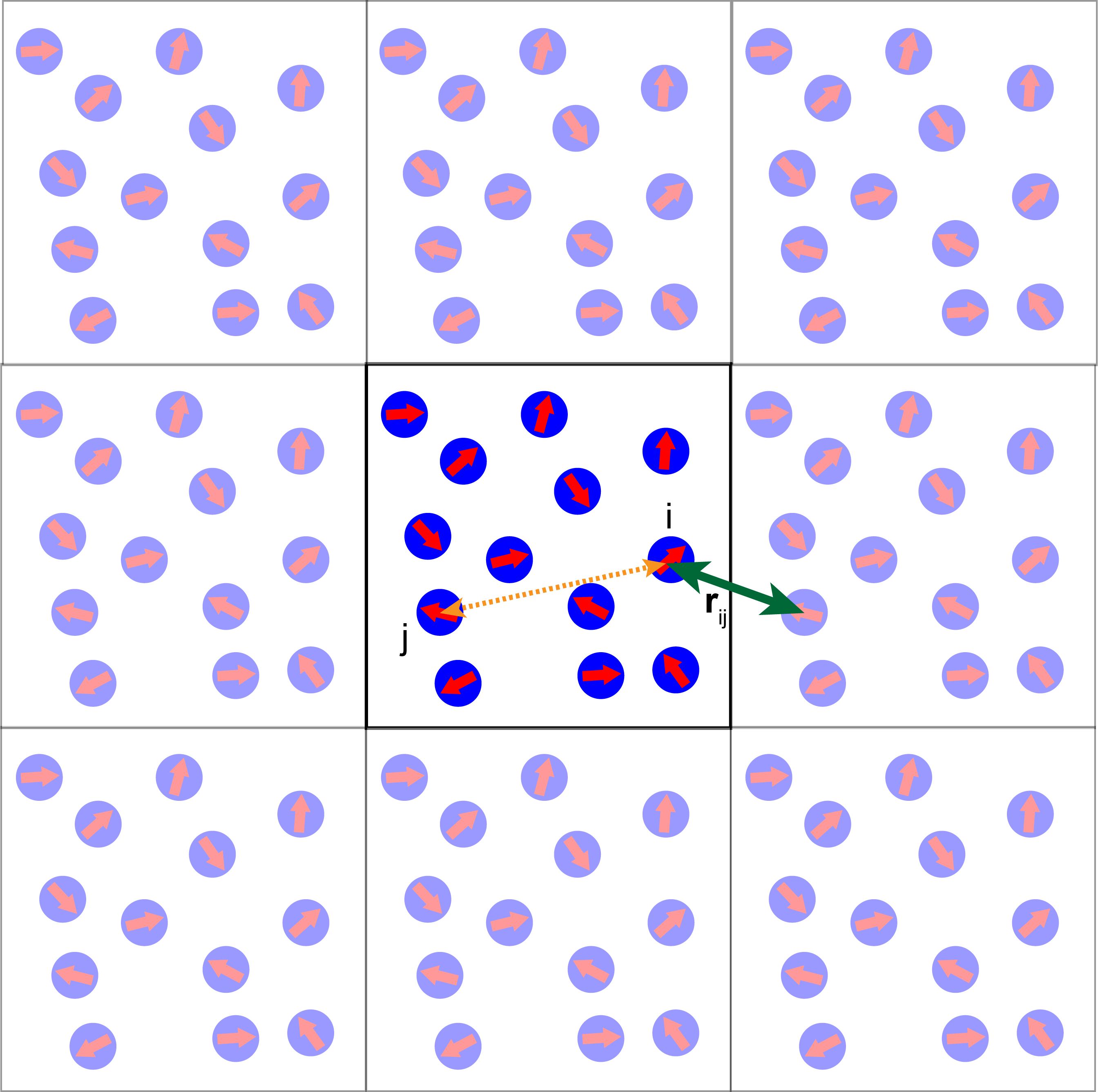
The idea behind PBC is that the simulation box is assumed to have the topology of a torus. That is, a particle that leaves the simulation box through the left boundary would instantaneously reappear on the right side of the simulation box. This implies that when computing the distance between two particles one has to consider all of their periodic images and pick the shortest distance. This is called the minimum image convention.
The idea behind the minimum image convention is shown in the image below.
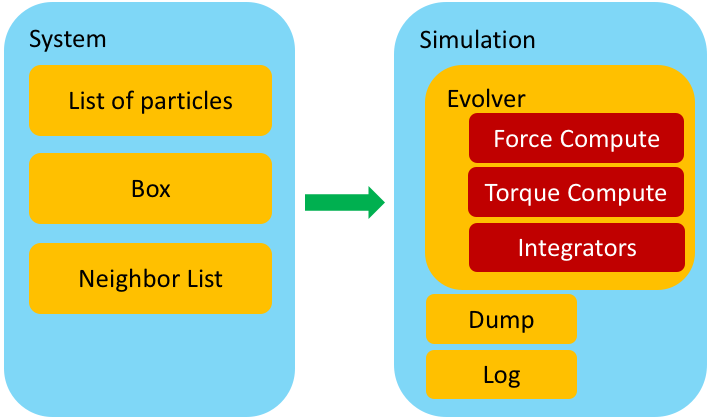
Key components of a particle-based simulation code¶
The image below shows the key components of a particle-based code and a possible design layout of how to organize them.
A working example¶
Let us start with a working example of a full simulation.
We read the initial configuration stored in the file init.json with $N=1,000$ randomly placed particles in a square box of size $L=50$. We assume that all particles have the same radius $a=1$. Further, each particle is self-propelled with the active force of magnitude $\alpha=1$ and experiences translational friction with friction coefficient $\gamma_t = 1$. Rotational friction is set to $\gamma_r = 1$ and the rotational diffusion constant to $D_r = 0.1$. Particles within the distance $d=2$ of each other experience the polar alignment torque of magnitude $J=1$.
We use the time step $\delta t = 0.01$ and run the simulation for $1,000$ time steps. We record a snapshot of the simulation once every 10 time steps.
from pymd.md import * # Import the md module from the pymd package
s = System(rcut = 3.0, pad = 0.5) # Create a system object with neighbour list cutoff rcut = 3.0 and padding distance 0.5
s.read_init('init.json') # Read in the initial configuration
e = Evolver(s) # Create a system evolver object
d = Dump(s) # Create a dump object
hf = HarmonicForce(s, 10.0, 2.0) # Create pairwise repulsive interactions with the spring contant k = 10 and range a = 2.0
sp = SelfPropulsion(s, 1.0) # Create self-propulsion, self-propulsion strength alpha = 1.0
pa = PolarAlign(s, 1.0, 2.0) # Create pairwise polar alignment with alignment strength J = 1.0 and range a = 2.0
pos_integ = BrownianIntegrator(s, T = 0.0, gamma = 1.0) # Integrator for updating particle position, friction gamma = 1.0 and no thermal noise
rot_integ = BrownianRotIntegrator(s, T = 0.1, gamma = 1.0) # Integrator for updating particle oriantation, friction gamma = 1.0, "rotation" T = 0.1, D_r = 0.0
# Register all forces, torques and integrators with the evolver object
e.add_force(hf)
e.add_force(sp)
e.add_torque(pa)
e.add_integrator(pos_integ)
e.add_integrator(rot_integ)
dt = 0.01 # Simulation time step
# Run simulation for 1,000 time steps (total simulation time is 10 time units)
for t in range(1000):
print("Time step : ", t)
e.evolve(dt) # Evolve the system by one time step of length dt
if t % 10 == 0: # Produce snapshot of the simulation once every 10 time steps
d.dump_vtp('test_{:05d}.vtp'.format(t))
A note on units¶
Step by step guide through the working example¶
Let us now dissect the example above line by line and look into what is going on under the hood.
In order to use the pymd package we need to load it. In particular, we import the md module.
from pymd.md import *
The source code on the entire module can be found in the ABPtutotial/Python directory.
Creating the System object¶
In the following line, we crate an instance of the System class. The System class stores the system we are simulating and the information about the simulation box. It is also in charge of reading the initial configuration from a JSON file and it also builds and updates the neighbor list.
s = System(rcut = 3.0, pad = 0.5) # Create a system object with neighbour list cutoff rcut = 3.0 and padding distance 0.5
s.read_init('init.json') # Read in the initial configuration
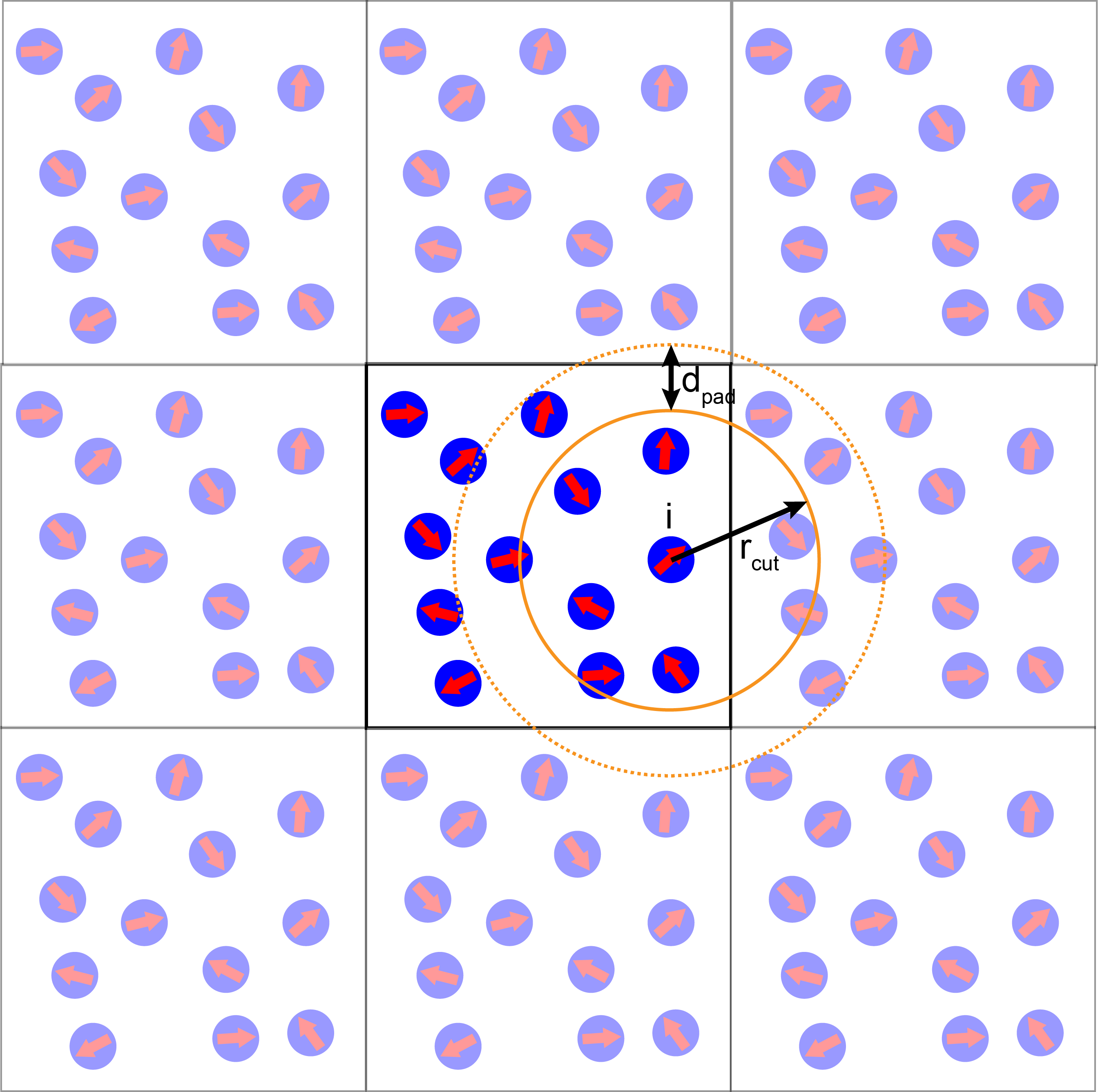
In our example, the instance of the System class is created with the neighbor list cutoff set to $r_{cut}=3.0$ and padding distance $d_{pad}=0.5$. For the system parameters used below, this corresponds approximately to 10 steps between neighbor list rebuilds.
After the System object has been created, we call the read_init member function to read in the size of the simulation box and the initial location and direction of the polarity vector for each particle.
System class is a part of the pymd.md core.
A note on neighbor lists¶
Below is a visual representation of a neighbor list:
The Evolver class¶
Now that we have created the System object and populated it with the initial configuration, we proceed to create the Evolver object:
e = Evolver(s) # Creates an evolver object
The Evolver class is the workhorse of our toy simulation. It's central member function is evolve which performs the one step of the simulation. Its task is to:
- Ensure that the neighbor list is up to date;
- Perform the integration pre-step;
- Compute all forces and torques on each particle;
- Perform the integration post-step;
- Apply periodic boundary conditions.
In our design, one can have multiple force and torque laws apply in the system. In addition, multiple integrators can act in the same system. More about what this means below.
Evolver class is a part of the pymd.md core.
The Dump class¶
The Dump class handles output of the simulation run. This is the most important part of the code for interacting with the postprocessing analysis tools. It takes snapshots of the state of the system at a given instance in time and saves it to a file of a certain type. Here, we support outputs in JSON and VTP formats.
Dump class is a part of the pymd.md core.
d = Dump(s) # Create a dump object
Forces and torques¶
In our implementation, each non-stochastic term in equations of motion is implemented as a separate class. Terms in the equation of motion for particle's position are all given a generic name force while terms that contribute to the orientation of particle's polarity are called torque. Note particles in our model are disks and that rotating a disk will not stir the system, so the term torque is a bit loose.
All forces and torques are stored in two lists in the Evolver class. The evolve member function of the Evolver class ensured that in each time step forces and torques on every particle are set to zero, before they are recomputed term by term.
While this design introduces some performance overhead (e.g., distances between particles have to be compute multiple times in the same time step), it provides great level flexibility since adding an additional term to the equations of motion requires just implementing a new Force class and adding it to the Evolver's list of forces.
These classes are defined in the forces and torques directories.
hf = HarmonicForce(s, 10.0, 2.0) # Create pairwise repulsive interactions with spring contant k = 10 and range 2.0
sp = SelfPropulsion(s, 1.0) # Create self-propulsion, self-propulsion strength alpha = 1.0
pa = PolarAlign(s, 1.0, 2.0) # Create pairwise polar alignment with alignment strength J = 1.0 and range 2.0
# Register all forces, torques and integrators with the evolver object
e.add_force(hf)
e.add_force(sp)
e.add_torque(pa)
Integrators¶
Finally, the equations of motion are solved numerically using a discretization scheme. This is done by a set of classes that we call Integrator classes. Each of these classes implement one of the standard discretization schemed for solving a set of coupled ODEs. In the spirit of our modular design, solver of each equation of motion is implemented as a separate Integrator class allowing the code to be modular and simple to maintain.
In our example, both equations of motion are overdamped and not stiff. However, both equations of motion contain stochastic terms. Therefore, in both cases we implement the simple first-order Euler-Maruyama scheme.
For the equation of motion for particle's position with time step $\delta t$, the discretization scheme (e.g., for the $x-$component of the position of particle $i$) is:
$x_i(t+\delta t) = x_i(t) + \delta t\left(v_0 n_{i,x} - \frac{1}{\gamma_t}\sum_j k\left(2a - r_{ij}\right)\frac{x_i}{r_{ij}} \right) + \sqrt{2\frac{T}{\gamma_r}\delta t}\mathcal{N}(0,1)$,
where $\mathcal{N}(0,1)$ is a random number drawn from a Gaussian distribution with the zero mean and unit variance. This integrator is implemented in the BrownianIntegrator class.
The implementation for the direction of the particle's orientation is similar and is implemented in the BrownianRotIntegrator class. Note that, for convenience, in this class we also introduced a parameter $T$, such that the rotation diffusion coefficient is defined as $D_r = \frac{T}{\gamma_r}$.
A note on numerical integration of equation of motion¶
pos_integ = BrownianIntegrator(s, T = 0.0, gamma = 1.0) # Integrator for updating particle position, friction gamma = 1.0 and no thermal noise
rot_integ = BrownianRotIntegrator(s, T = 0.1, gamma = 1.0) # Integrator for updating particle oriantation, friction gamma = 1.0, "rotation" T = 0.1, D_r = 0.0
# add the two integrtors to the Evolver's list of integrtors
e.add_integrator(pos_integ)
e.add_integrator(rot_integ)
Iterating over time¶
Now that we have all ingredients in place, we can run a simulation. In our implementation, this is just a simple "for" loop over the predefined set of time steps. Inside this loop we perform the basic data collection, e.g., saving snapshots of the state of the system.
dt = 0.01 # Simulation time step
# Run simulation for 1,000 time steps (total simulation time is 10 time units)
for t in range(1000):
print("Time step : ", t)
e.evolve(dt) # Evolve the system by one time step of length dt
if t % 10 == 0: # Produce snapshot of the simulation once every 10 time steps
d.dump_vtp('test_{:05d}.vtp'.format(t))
Final remarks on implementation¶
In the case of a full production-grade code, it would be convenient to add several more components. For example:
Logger classes would be in charge of handling logging of various observables that can be compute on "fly", such as the total energy of the system (arguably, of not much use in an active, self-propelled system), pressure, stress, etc.
Event classes would be reporting key events, such as reporting if the new force term was added, what is the frequency of saving the data, etc.
It would also be useful to save the entire state of the simulation (i.e., all parameters for each particle and all forces, torques and integrators) for future restarts.
One of the key parts of any good code, which is, unfortunately, often overlooked is having a set of automatic tests. As the code grows in complexity, making sure that adding new features does not break the expected behavior of the existing components becomes a challenging task. A modular design that decouples different parts of the code from each other as much as possible, together with a powerful set of automatic tests is essential to make this task tractable.
Visualizing results¶
There are many excellent tools for visualizing the results of a particle-based simulation. Commonly used are VMD, PyMol and Chimera, to name a few. In this tutorial we use Paraview.
Analyzing results of a simulation¶
This is the third step in the simulation workflow. Once we have collected particle trajectories it is time to extract relevant information about the system. In this step we learn something new about the system we study and, in many ways, this the key and most creative step in doing simulation-based research. Addressing the the analysis of simulation results could be a subject of a long series of tutorials.